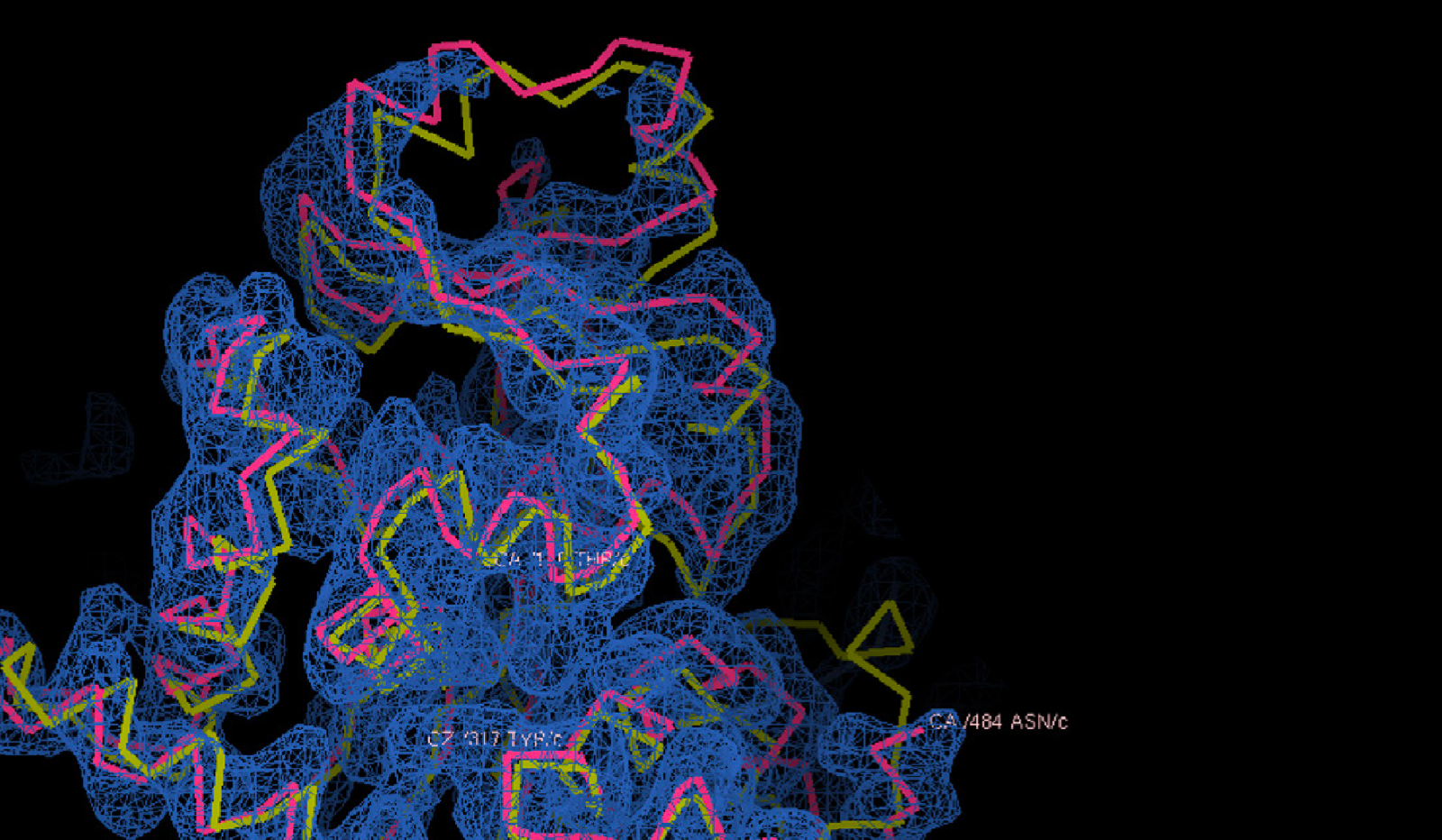
Confronto tra la struttura prevista da AlphaFold2, in giallo, e quella determinata sperimentalmente tramite microscopia crioelettronica, reticolo blue, della proteina gp105, una subunità dell'RNA polimerasi del batteriofago AR9. Fonte: presentazione di Petr Leiman, University of Texas Medical Branch, alla conferenza di presentazione dei risultati di CASP14.
"Questo cambierà tutto". "L'intelligenza artificiale trionfa nel calcolo della struttura delle proteine". "DeepMind ha risolto una sfida della biologia lunga 50 anni". "La soluzione di un mistero della biologia". "Uno dei più grandi misteri della biologia sostanzialmente risolto da un'intelligenza artificiale". Con questi titoli i maggiori quotidiani del mondo hanno riportato la notizia che un sistema di intelligenza artificiale, sviluppato dalla società londinese DeepMind, è stata in grado di determinare la struttura tridimensionale di un centinaio di proteine con un'accuratezza mai raggiunta prima e paragonabile a quella delle tecniche sperimentali che finora hanno svelato la maggior parte delle strutture proteiche che conosciamo.
La notizia è arrivata dagli organizzatori della competizione CASP (Critical Assessment of techniques for protein Structure Prediction), che si tiene ogni due anni e che quest'anno è giunta alla sua quattordicesima edizione. Dal 1994 CASP organizza una sfida tra decine di gruppi di ricerca nel campo della biologia computazionale chiedendogli di prevedere la struttura tridimensionale di un campione di proteine e complessi proteici a partire dalla sequenza di amminoacidi che li costituiscono. Quest'anno AlphaFold2, questo il nome dell'intelligenza artificiale sviluppata da DeepMind, ha vinto di misura su tutti gli altri partecipanti.
La struttura tridimensionale delle proteine stabilisce il loro funzionamento e di conseguenza regola alcune attività fondamentali all'interno delle cellule degli organismi viventi. La conoscenza del modo in cui la catena di amminoacidi che costituisce ciascuna proteina si distribuisce nello spazio, come i tratti di questa catena si torcano su se stessi a formare delle strutture elicoidali o si ripieghino a disegnare bracci che si diramano dall'asse centrale, è cruciale per capire i meccanismi alla base di molte malattie e per progettare farmaci efficaci. Anche per la pandemia in corso la conoscenza della forma delle proteine che concorrono alle diverse fasi del ciclo di vita del virus SARS-CoV-2 all'interno della cellula ospite che ha infettato viene utilizzata per la ricerca e il riposizionamento di antivirali efficaci contro la COVID-19. Grazie alla cristallografia a raggi X e, più recentemente alla criomicroscopia elettronica, conosciamo oggi la struttura di circa 170 mila proteine, che però costituiscono solo il 17% dell'insieme delle proteine presenti nel corpo umano. Questi metodi sperimentali sono infatti costosi e richiedono tempo.
Per questo motivo da circa cinquanta anni gli scienziati hanno tentato di risolvere il problema insegnando ai computer a calcolare il modo in cui gli amminoacidi si dispongono in eliche e bracci partendo dalla sequenza della proteina, una stringa di lettere di lunghezza variabile (da qualche decina a qualche centinaio, se si considerano i domini ovvero le porzioni di proteine che possono essere studiate individualmente). Ciascuna lettera di questa stringa può assumere uno tra 21 valori, codificati in una lettera dell'alfabeto: A per Alanina, C per la Cisteina, D per l'acido aspartico, e così via fino alla Y di Tirosina. Hanno cominciato quando il biochimico Christian Anfinsen vinse il Nobel per la chimica nel 1972 dimostrando che le sequenze di amminoacidi determinano completamente la struttura delle proteine, ma i primi tentativi furono fallimentari.
Calcolare la struttura da principi primi è infatti impossibile. Ciascuna proteina è costituita da centinaia di amminoacidi e ciascuno di essi, a sua volta, contiene una decina di atomi, e come questi si dispongano nello spazio dipende dalle interazioni tra di essi stabilite dalle leggi della fisica. I metodi computazionali si sono quindi orientati verso la ricerca di somiglianze tra le sequenze di proteine dalla struttura nota per provare a dedurre, statisticamente, le somiglianze tra le strutture associate. La competizione CASP ha cercato di accelerare il progresso in questa area creando ed accrescendo nel tempo un database di strutture proteiche note dagli esperimenti che potessero essere usate come riferimento e stabilendo delle metriche di confronto tra i vari modelli che via via sono stati sviluppati.
Ogni due anni il comitato scientifico di CASP rende note le sequenze di un campione di circa cento tra proteine e domini la cui struttura è stata determinata sperimentalmente ma viene tenuta segreta per tutta la durata della gara. Nell'arco di alcuni mesi i partecipanti alla competizione calcolano con i loro modelli le strutture proteiche e le inviano ai giudici del concorso per la valutazione. Ciascuna struttura viene confrontata con quella sperimentale, la cosiddetta ground truth, e riceve un punteggio da 0 a 100, chiamato Global Distance Test Total Score (GDT_TS). Il punteggio viene calcolato partendo dalle percentuali di amminoacidi che, rispetto alla struttura sperimentale, si trovano a distanze inferiori a 1, 2, 4 e 8 Angstrom (una lunghezza pari a un decimo di nanometro, cioè alla dimensione di un atomo). Queste quattro percentuali vengono mediate e così si ottiene il GDT_TS. Quanto più il GDT_TS è vicino al 100%, tanto più la struttura calcolata è vicina a quella osservata negli esperimenti. Questo score viene poi mediato su tutte le strutture del campione per stabilire la performance media del gruppo e, dopo una procedura di standardizzazione, confrontato con i punteggi ottenuti da tutti gli altri gruppi per stilare la classifica finale.
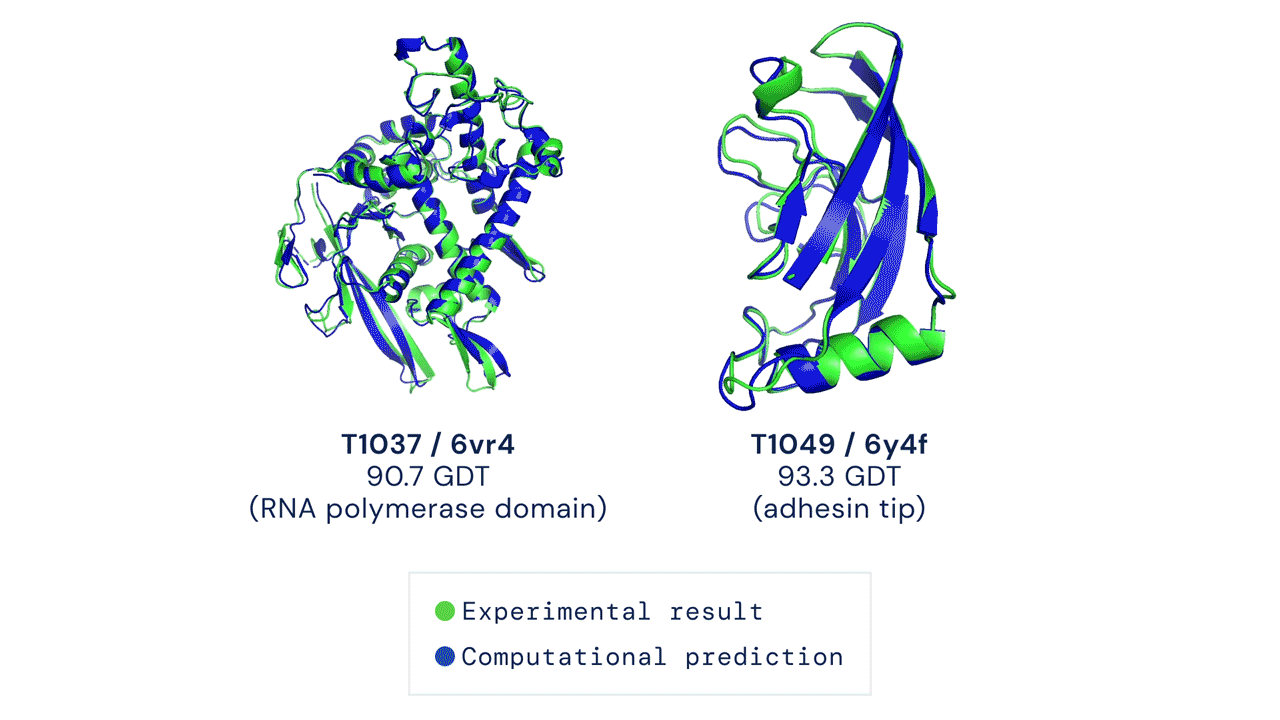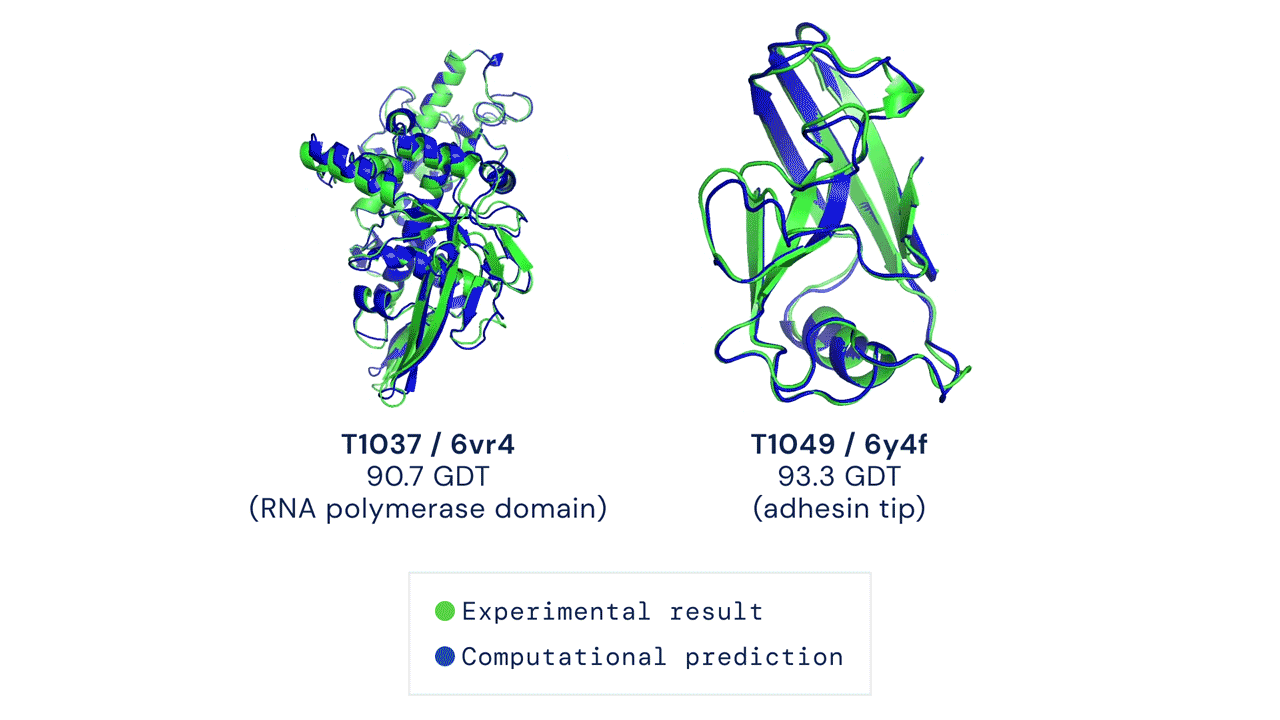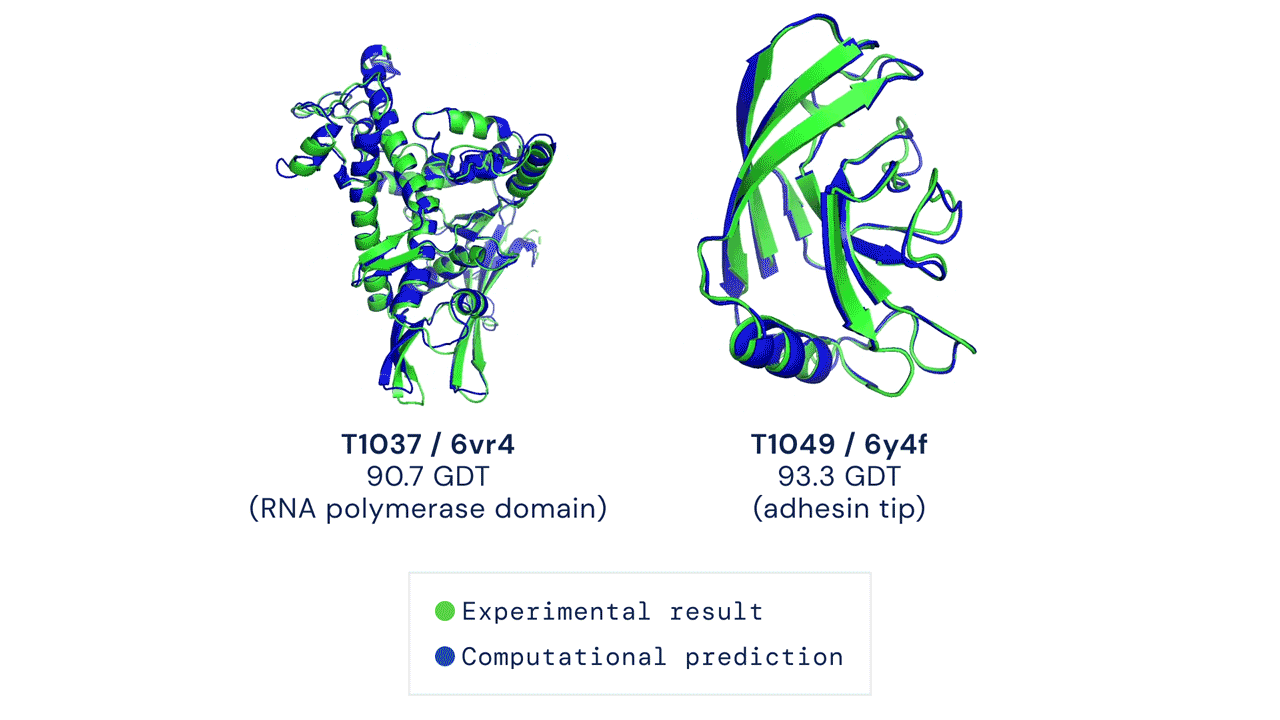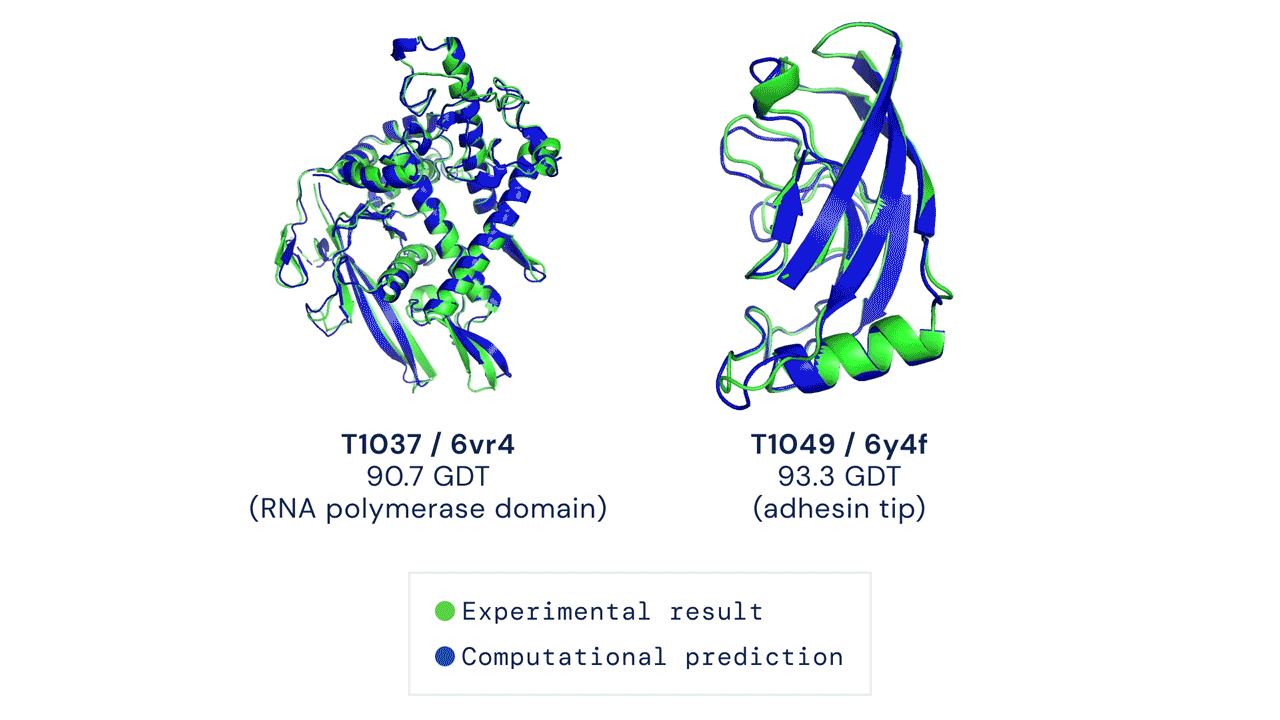
Due esempi di proteine nel campione free modelling della competizione CASP14. Confronto tra la struttura prevista da AlphaFold2 (in blu) e quella sperimentale (in verde). Credit: DeepMind via PhysOrg.
Quest'anno l'algoritmo AlphaFold2 ha ottenuto un punteggio medio di 92,4 e per il sottoinsieme di proteine considerate più difficili un punteggio medio di 87, circa 25 punti sopra il secondo gruppo classificato, quello guidato da David Baker della University of Washington. Due terzi delle strutture calcolate da AlphaFold2 hanno ottenuto un punteggio superiore a 90. Questi punteggi possono essere tradotti in termini di RMSD (root mean square deviation) che misura la differenza fra le coordinate atomiche del modello computazionale e quelle della struttura sperimentale. Per AlphaFold2 il valore di RMSD è pari a 1,6 Angstrom. Questo può essere confrontato con quello ottenuto sperimentalmente, che nel caso delle strutture a risoluzione media è di 0,6 Angstrom e può arrivare fino a 0,1-0,2 Angstrom.
«Questi sono gli ordini di grandezza necessari nelle routine di drug discovery, ma visto il salto di accuratezza fatto rispetto al 2018 non sembra improbabile che questi sistemi possano raggiungerli nel prossimo futuro», commenta Silvia Onesti, che dirige la divisione di biologia strutturale del sincrotrone Elettra a Trieste, e aggiunge «il risultato di AlphaFold2 è sorprendente, ma più che vederlo come uno strumento che sostituirà la cristallografia delle proteine, credo possa diventare sinergico. Il lavoro dei biologi strutturali oggi va oltre la ricostruzione della forma delle proteine a partire dai pattern di diffrazione dei raggi X. I ricercatori del mio gruppo dedicano una considerevole quantità di tempo alla produzione e purificazione delle proteine e queste vengono usate solo in parte per gli studi di cristallografia. Un'altra parte viene sfruttata per indagare le loro proprietà biochimiche». La tecnica che per ora sembra meno minacciata dagli avanzamenti di AlphaFold2 è senz'altro quella della microscopia crioelettronica, specializzata nella risoluzione della struttura di grandi complessi proteici. AlphaFold2, infatti, è in grado di calcolare la struttura di proteine che contengono fino ad alcune centinaia di amminoacidi, mentre molti complessi macromolecolari che hanno un ruolo importante in biologia possono contenerne alcune migliaia.
Il risultato di AlphaFold2 rappresenta comunque un enorme passo avanti rispetto alla scorsa edizione di CASP, quella del 2018, la prima a cui DeepMind ha preso parte con l'algoritmo AlphaFold, realizzando un punteggio medio poco sotto 60, e posizionandosi circa 6 punti sopra il secondo classificato. «Pensavo avremmo dovuto aspettare dieci anni per arrivare dai risultati di AlphaFold del 2018 a quelli di quest'anno. Siamo vicini al limite fisico dell'accuratezza. Fondamentalmente queste strutture sono flessibili. Non ha senso parlare di risoluzioni superiori», ha dichiarato a MIT Technology Review Mohammed AlQuraish, biologo computazionale della Columbia University e uno dei partecipanti alla competizione.
AlphaFold2 ha stracciato tutti gli avversarsi anche secondo un'altra metrica, che considera distanze più piccole rispetto al punteggio che abbiamo descritto prima e serve a valutare l'accuratezza della struttura in maniera ancora più stringente. Andrei Lupas, biologo evoluzionista del Max Planck Institute for Developmental Biology a Tubinga in Germania, ha guidato la valutazione 'High Accuracy', presentando i risultati martedì 1 dicembre. Tuttavia, entrambe queste metriche misurano l'accuratezza globale della struttura e non quella locale, come spiega AlQuraish in un editoriale del 2019 sulla rivista Bioinformatics, commentando i risultati di AlphaFold nella scorsa edizione di CASP: «l'accuratezza a livello locale relativa, per esempio al coordinamento nel movimento degli atomi in un sito attivo o al cambiamento localizzato di conformazione dovuta a una mutazione, può essere l'aspetto più importante di una struttura per rispondere a un insieme più ampio di domande biologiche».I risultati di AlphaFold2 restano sorprendenti, quasi incredibili, così Lupas ha voluto testarli ulteriormente, sottoponendogli la sequenza di una proteina studiata nel suo laboratorio che, nonostante la disponibilità dei dati di diffrazione dai raggi X, non era riuscito a risolvere. Grazie alla struttura calcolata da AlphaFold2, i ricercatori del gruppo di Lupas hanno ottenuto un modello computazionale della loro proteina che gli ha permesso di fasare e interpretare i dati di diffrazione: «È praticamente perfetta, non ho idea di come ci riescano», ha detto a Science.
Per ora nessuno sa come DeepMind sia riuscito a ottenere questi risultati sorprendenti. Demis Hassabis, CEO della società, ha dichiarato che i dettagli sul funzionamento di AlphaFold2 saranno resi noti in un articolo scientifico che probabilmente verrà pubblicato il prossimo anno. Martedì, secondo quanto riportato sul programma della conferenza, John Jumper, team leader di AlphaFold2, ha presentato i risultati della sua squadra e potrebbe aver dato qualche dettaglio in più, ma finora le sue slides non sono state archiviate insieme a quelle degli altri gruppi. Qualche informazione ci arriva dalle dichiarazioni di Jumper e dall'abstract pubblicato qui. Il cuore dell'algoritmo è una rete neurale profonda con un'architettura nuova, chiamata attention based, capace di concentrarsi su porzioni più piccole della struttura proteica e poi assemblare ciò che ha appreso per prevedere la forma completa, come si farebbe con i pezzi di un puzzle.
Tuttavia, pare che le differenze fra AlphaFold2 e AlphaFold siano limitate alla componente di deep learning dell'algoritmo, che è solo una parte dell'intero sistema. Possiamo, dunque, riferirci ad AlphaFold per parlare dell'impostazione generale del problema. La struttura della proteina viene schematizzata come una catena di amminoacidi e la sua geometria viene descritta dalle distanze tra ciascuna coppia di amminoacidi (2L variabili se L è la lunghezza della sequenza) e una coppia di angoli di torsione per ciascun amminoacido (4L variabili in tutto). AlphaFold è composto di tre passi. Nel primo si estraggono le caratteristiche delle sequenze di amminoacidi che sono rilevanti per la struttura finale assunta dalla proteina. Questo si fa cercando nel database su cui si svolge l'apprendimento, costituito dalle 170 mila proteine contenute nel Protein Data Bank, delle altre sequenze che hanno porzioni simili. Si costruisce così un insieme di omologhi della proteina che si vuole studiare, insieme che condivide un antenato comune. Ricostruendo il processo di evoluzione, si è in grado di codificare le informazioni rilevanti per la struttura della proteina che sono contenute nella sua sequenza genetica. Nel secondo passo queste informazioni vengono date in pasto a una rete neurale profonda che le utilizza per prevedere le distribuzioni probabilistiche delle distanze tra tutte le coppie di amminoacidi presenti nella sequenza e dei loro angoli di torsione. Le distribuzioni probabilistiche sono sostanzialmente degli istogrammi che dicono qual è la probabilità che la distanza tra due specifici amminoacidi sia contenuta in un certo intervallo di valori. Queste distribuzioni vengono utilizzate dal terzo passo dell'algoritmo, che calcola la posizione di ciascun amminoacido che meglio rispetti i vincoli stabiliti dalle distribuzioni su distanze e angoli. Questo calcolo è effettuato con metodi numerici convenzionali. La struttura teorica così ottenuta viene confrontata con quella sperimentale e, mediante un processo iterativo, i pesi della rete neurale vengono aggiustati per ottenere la migliore corrispondenza possibile tra le due.
Per AlphaFold era stata utilizzata una rete neurale con una architettura molto sofisticata, chiamata dilated residual convolutional neural network, descritta in dettaglio in questo articolo pubblicato su Nature all'inizio del 2020. Si stratta di una rete neurale molto profonda, con 220 strati interni e circa 21 milioni di parametri che descrivono le connessioni tra i nodi disposti sui diversi strati.
È importante sottolineare come il lavoro di DeepMind, prima con AlphaFold e poi con AlphaFold2, sia stato costruito su due decenni di progressi accumulati nel campo della biologica computazionale. Da una parte il metodo basato sulla ricerca degli omologhi e dei meccanismi di co-evoluzione, dall'altra i software che traducono le informazioni su distanze e angoli di torsione in strutture tridimensionali. DeepMind ha però portato questi strumenti su un'altra scala, raggiungendo risultati che solo due anni fa sarebbero stati impensabili. Inoltre, ha influenzato l'intero campo di ricerca: quest'anno circa la metà dei gruppi partecipanti ha impiegato qualche forma di deep learning nei suoi algoritmi.
Dame Janet Thornton, direttrice emerita dello European Bioinformatics Institute non coinvolta nella competizione CASP, ha dichiarato: «Cominciavo a pensare che non avrei visto la soluzione al problema nell'arco della mia vita. Una maggiore comprensione della struttura delle proteine e la possibilità di prevederle usando un computer, aprirà la strada a nuove scoperte nel campo dell'evoluzione e della medicina, come per esempio comprendere come variazioni genetiche osservate tra le persone siano legate all'insorgenza di alcune malattie».
Perché questo scenario si realizzi è necessario che AlphaFold2 venga condiviso con la comunità scientifica e che sia utilizzabile nella pratica dei laboratori. Demis Hassabis, il CEO di DeepMind, ha dichiarato che intende rendere AlphaFold2 accessibile a tutti i ricercatori. Per ora il training dell'algoritmo richiede alcune settimane usando il potere computazionale equivalente a quello di 100 o 200 GPU e impiega alcuni giorni per determinare la struttura di una singola proteina (ricordiamo che i complessi di proteine sono per ora fuori dalla sua portata).
Di certo AlphaFold2 segna un cambio di passo per DeepMind. La società londinese finora si era concentrata sulla soluzione di problemi matematicamente molto complessi, come il gioco degli scacchi o del Go, ma il suo obiettivo di lungo periodo è quello di sviluppare la cosiddetta artificial general intelligence. Non sappiamo se il risultato appena ottenuto sia un passo importante in quella direzione, ma di certo promette di avere un impatto enorme sul modo di fare ricerca nel campo della biologia e della medicina.
Per ricevere questo contenuto in anteprima ogni settimana insieme a sei consigli di lettura iscriviti alla newsletter di Scienza in rete curata da Chiara Sabelli (ecco il link per l'iscrizione). Trovi qui il testo completo di questa settimana.
Buona lettura, e buon fine settimana!


{kind=link}